

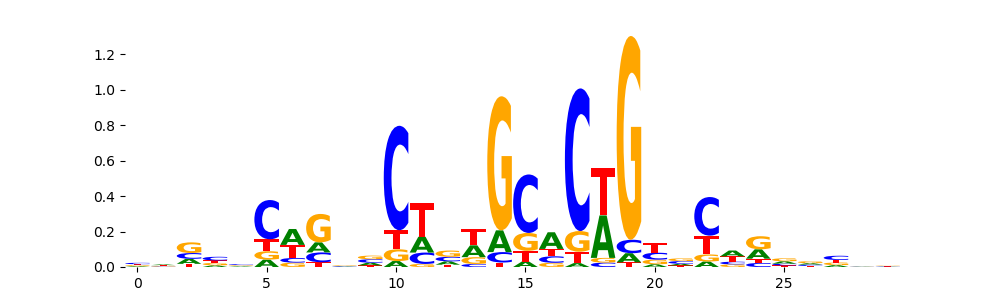
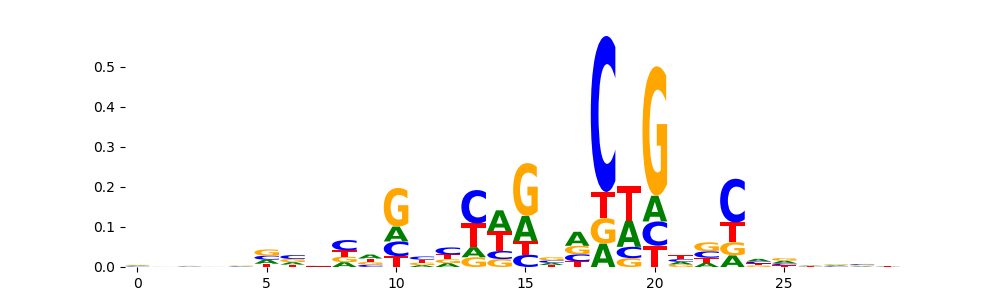
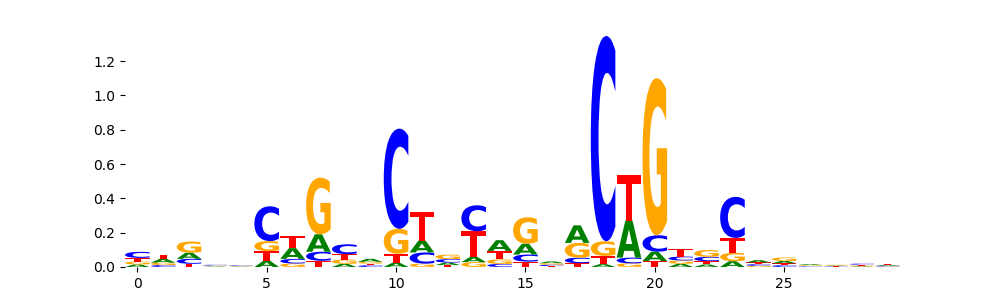


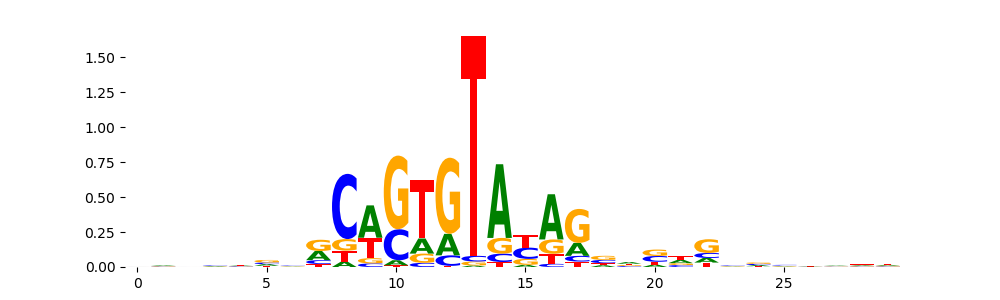
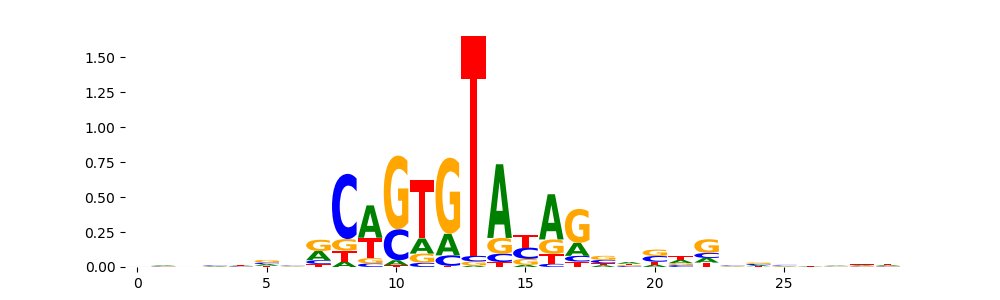
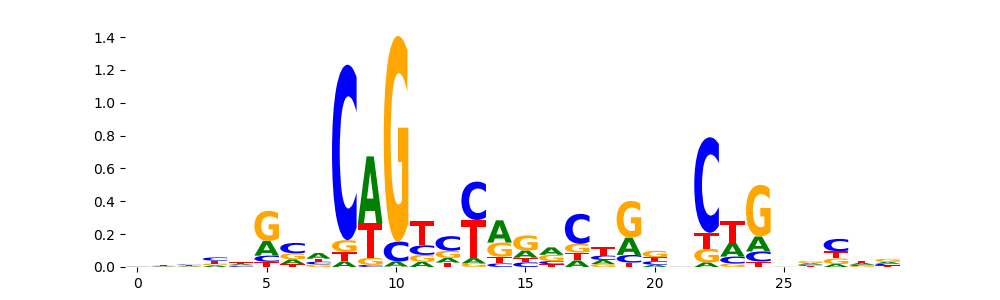


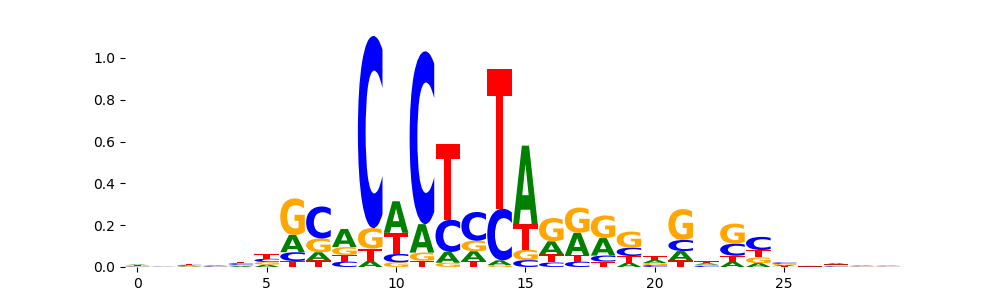




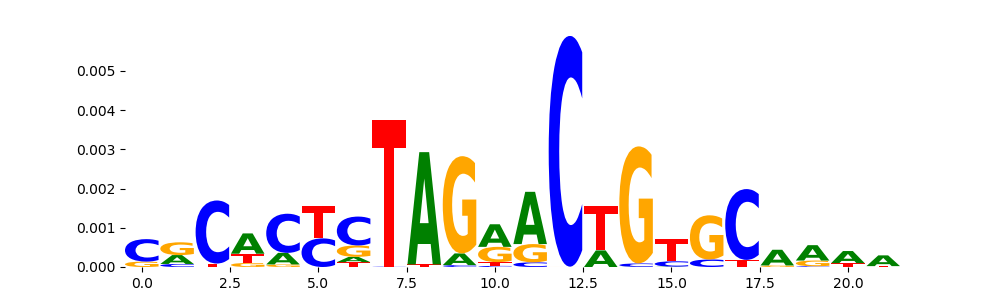
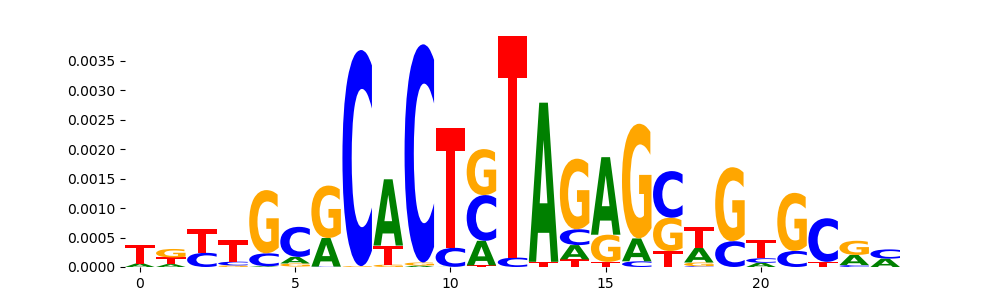




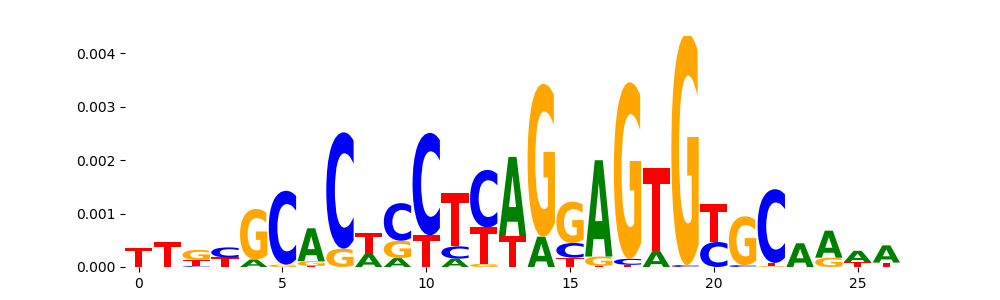
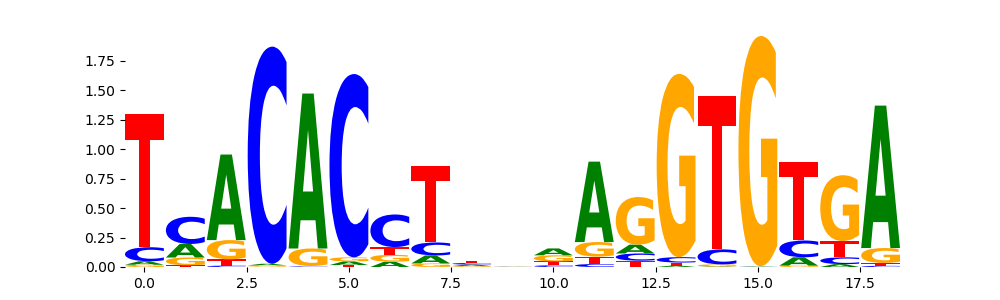






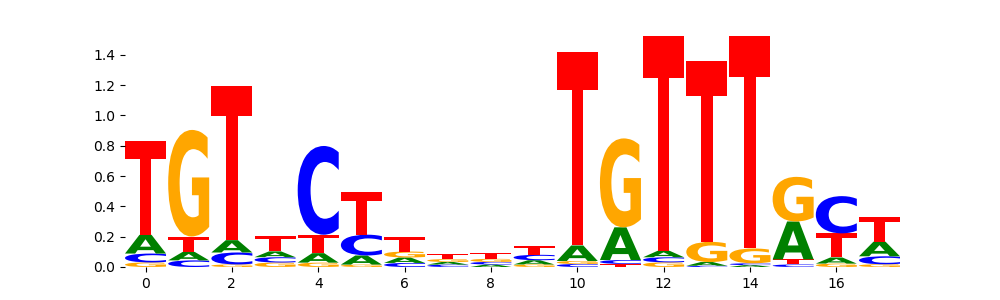
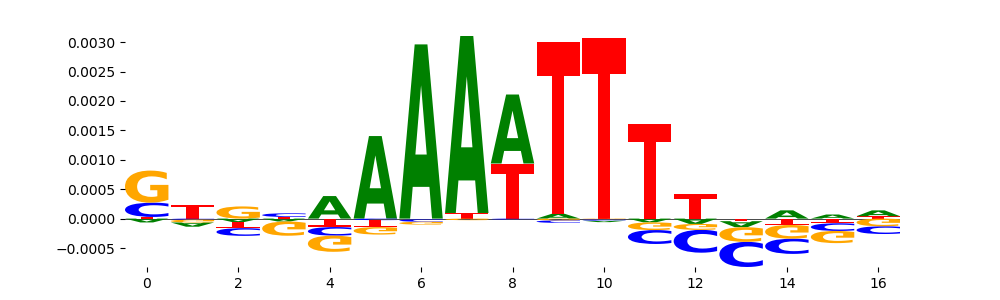
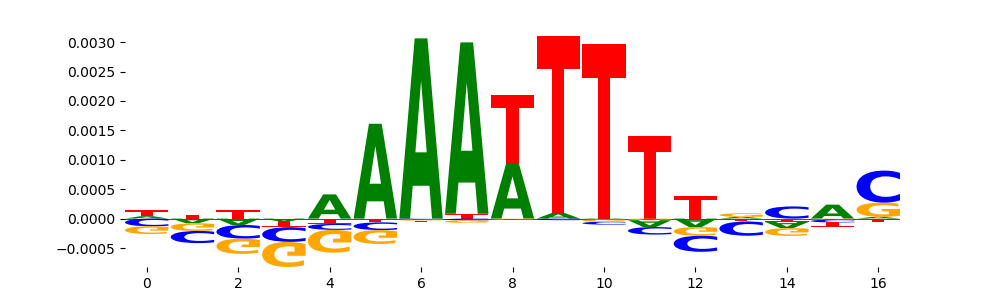
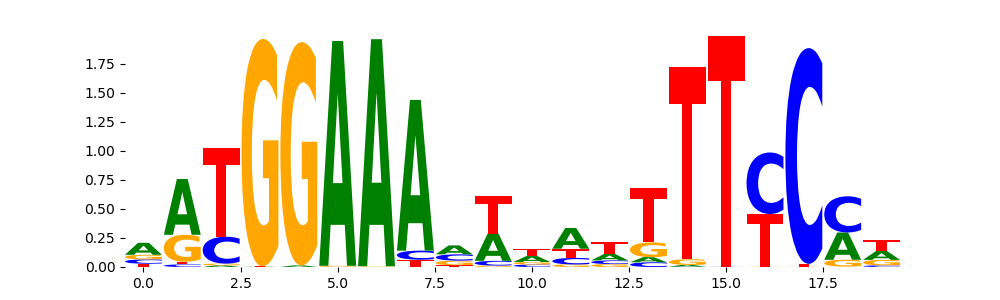
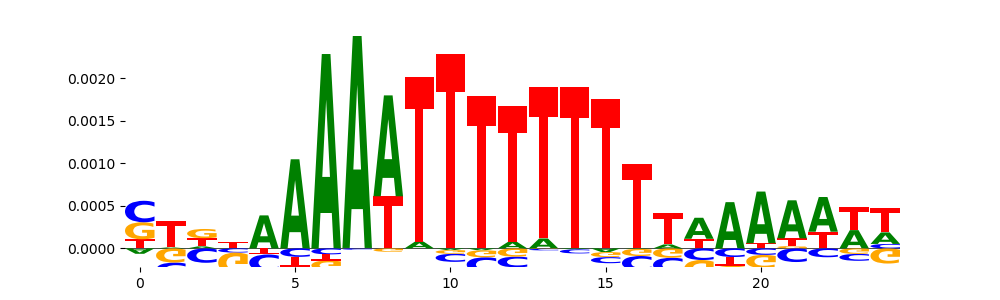


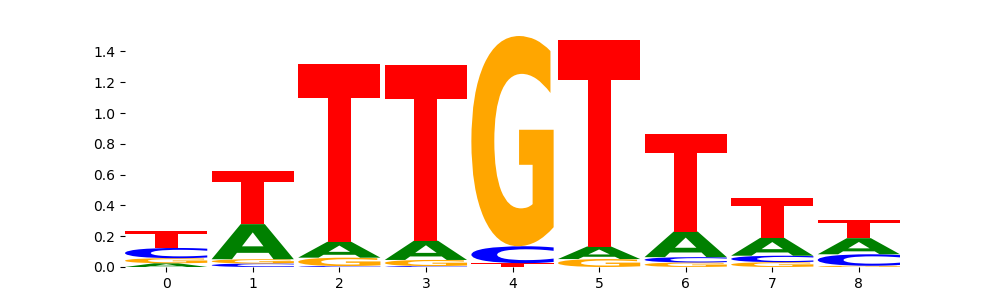





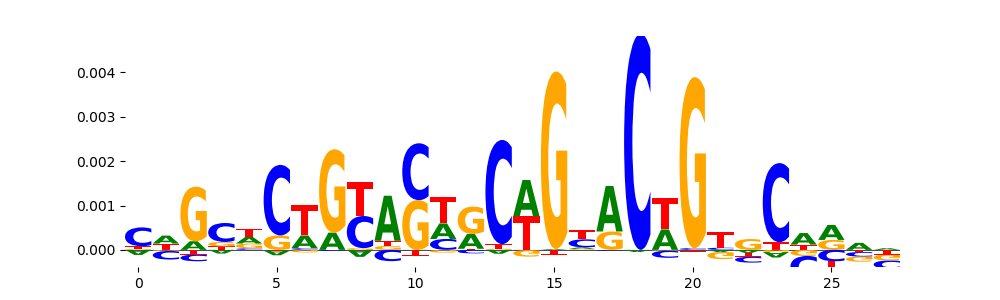
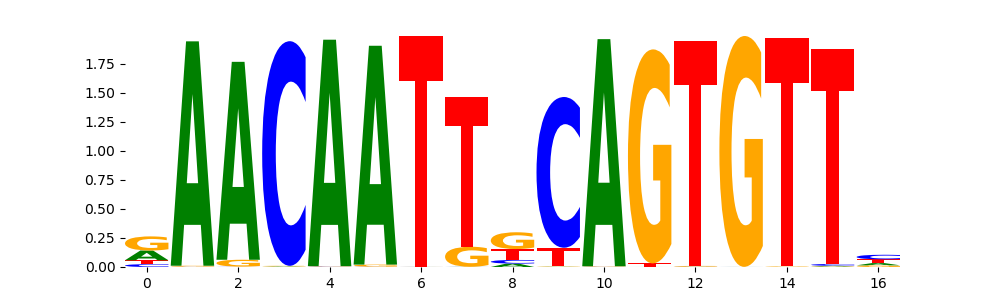

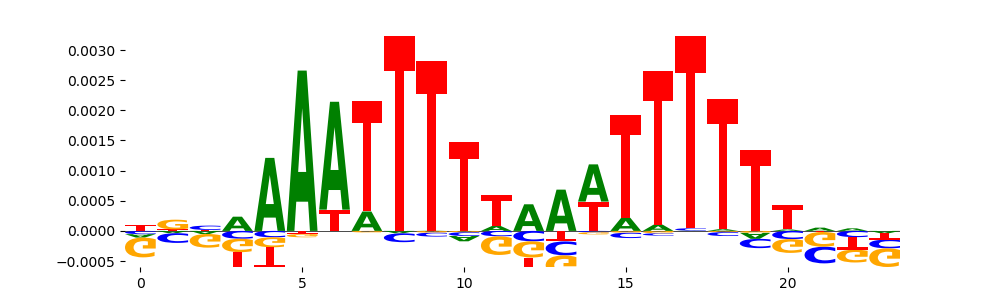

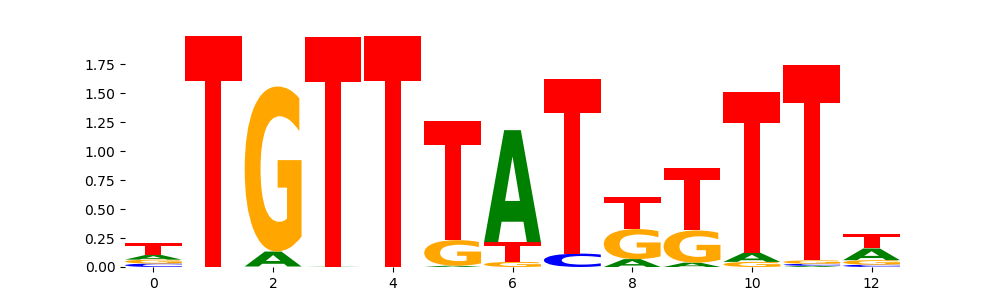
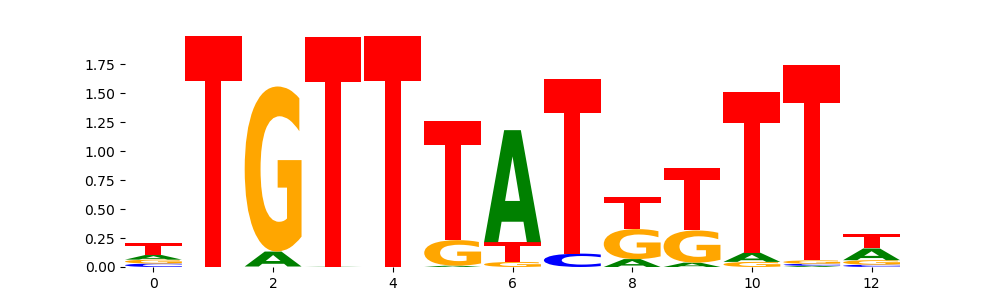

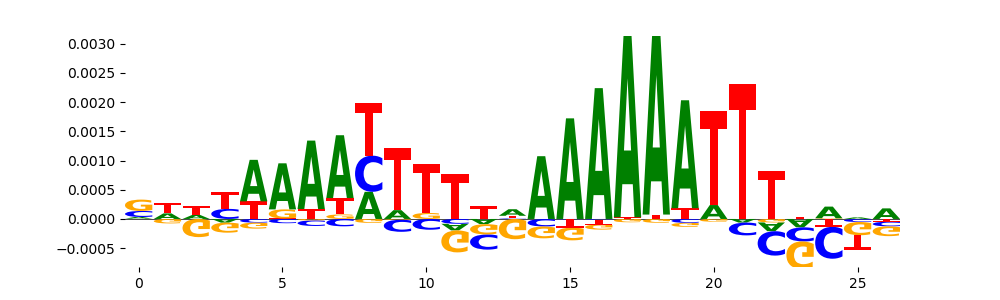
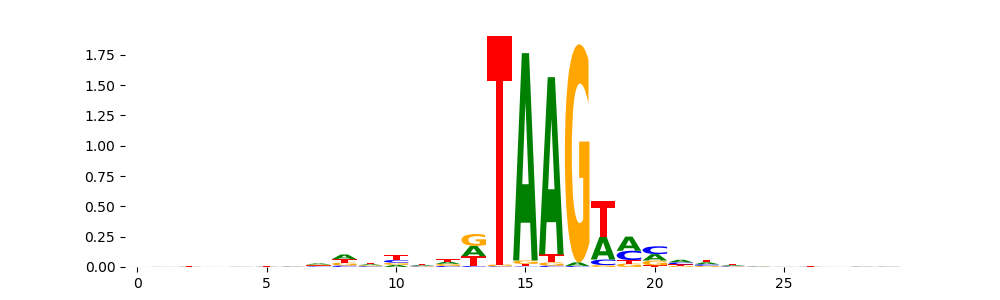
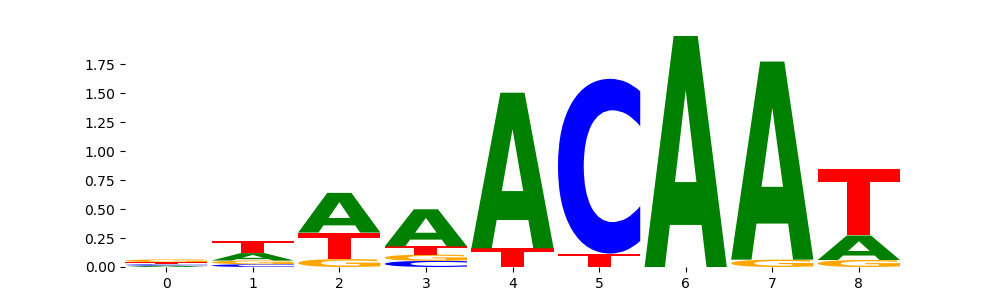
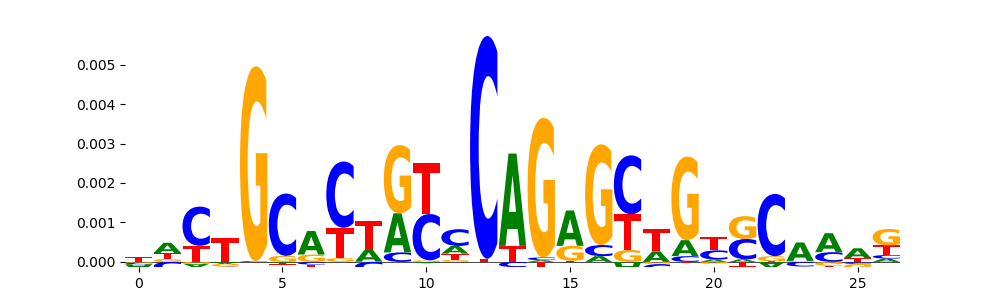

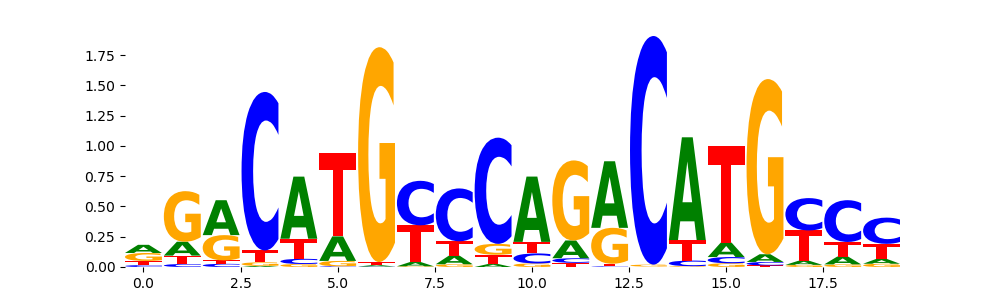
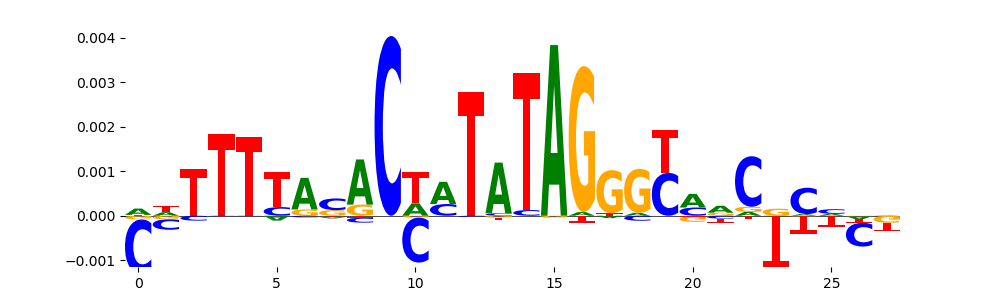




The image below should look closely like a Tn5 or DNase bias enzyme motif.
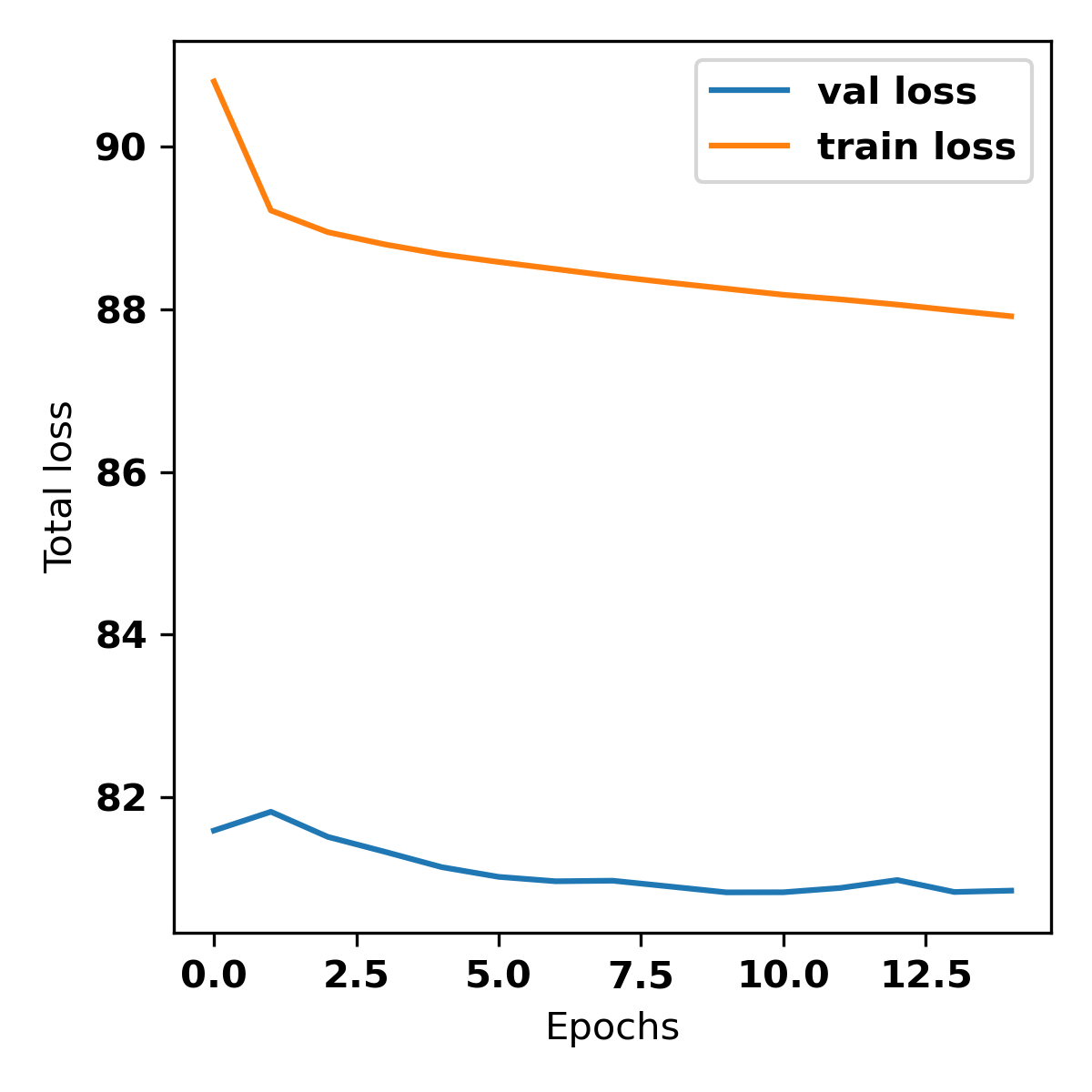
The val loss (validation loss) will decrease and saturate after a few epochs.
Counts Metrics: The pearsonr in non-peaks should be greater than 0 (higher the better). The pearsonr in peaks should be greater than -0.3 (otherwise the bias model could potentially be capturing AT bias). MSE (Mean Squared Error) will be high in peaks.
Profile Metrics: Median JSD (Jensen Shannon Divergence between observed and predicted) lower the better. Median norm JSD is median of the min-max normalized JSD where min JSD is the worst case JSD i.e JSD of observed with uniform profile and max JSD is the best case JSD i.e 0. Median norm JSD is higher the better. Both JSD and median norm JSD are sensitive to read-depth. Higher read-depth results in better metrics.
What to do if your pearsonr in peaks is less than -0.3? In the range of -0.3 to -0.5 please be wary of your chrombpnet_wo_bias.h5 (that wil potentially be trained with this bias model) TFModisco showing lots of GC rich motifs (> 3 in the top-10). If this is not the case you can continue using the chrombpnet_wo_bias.h5. If you end up seeing a lot of GC rich motifs it is likely that bias model has learnt a different GC distribution than your GC-content in peaks. You might benefit from increasing the bias_threshold_factor argument input to the chrombpnet bias pipeline or chrombpnet bias train command used in training the bias model and retrain a new bias model. For more intuition about this argument refer to the FAQ section in wiki. If the value is less than -0.5 the chrombpnet training will automatically throw an error.
| nonpeaks.pearsonr | nonpeaks.mse | peaks.pearsonr | peaks.mse | |
|---|---|---|---|---|
| counts_metrics | -0.2 | 2.66 | -0.45 | 12.12 |
| nonpeaks.median_jsd | nonpeaks.median_norm_jsd | peaks.median_jsd | peaks.median_norm_jsd | |
|---|---|---|---|---|
| profile_metrics | 0.64 | 0.15 | 0.5 | 0.25 |
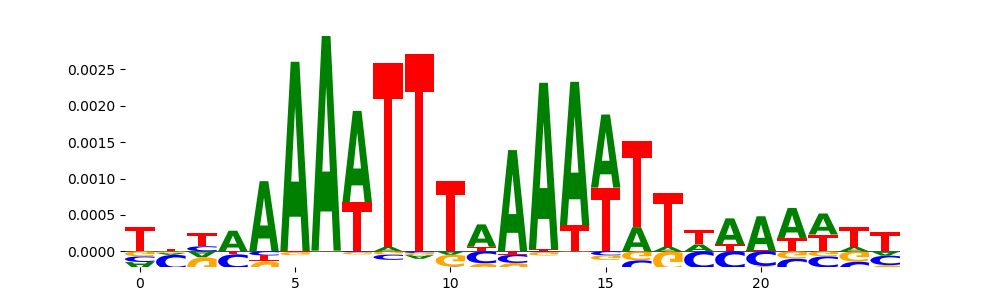
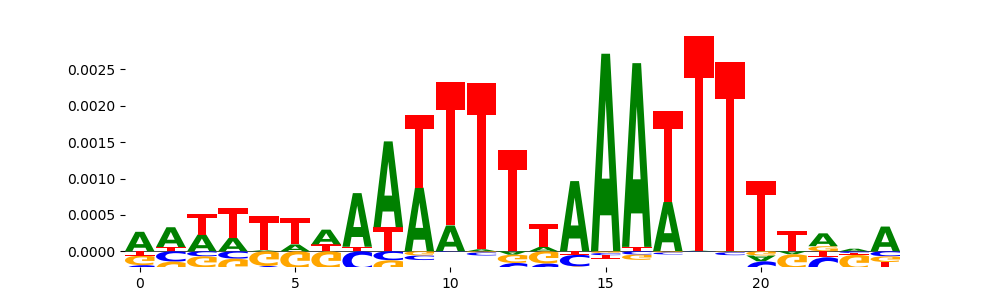
TFModisco motifs generated from profile contribution scores of the bias model. cwm_fwd, cwm_rev are the forward and reverse complemented consolidated motifs from contribution scores in subset of random peaks. These CWM motifs should be free from any Transcription Factor (TF) motifs and should contain either only bias motifs or random repeats. For each of these motifs, we use TOMTOM to find the top-3 closest matches (match_0, match_1, match_2) from a database consisting of both MEME TF motifs and heterogenous enzyme bias motifs that we have repeatedly seen in our datasets. The qvals (qval0,qval1,qval2) should be high (> 0.0001) if the closest hit is a TF motif (i.e indicating that the closest match is not the correct match) - this is also generally verifiable by eye as the closest match will look nothing like the CWMs. The qvals should be low if the closest hit is enzyme bias motif and generally verifiable that the top match looks like the CWM. The first 3-5 motifs in the list below should look like enzyme bias motif.
What to do if you find an obvious TF motif in the list?
Do not use this bias model as it will regress the contribution of the TF motifs (along with bias motifs) from the chrombpnet_nobias.h5. Reduce the bias_threshold_factor argument input to the chrombpnet bias pipeline or chrombpnet bias train command used in training the bias model and retrain a new bias model. For more intuition about this argument refer to the FAQ section in wiki.
What to do if you are unsure if a given CWM motif is resembling the match_0 logo for example?
Get marginal footprint on the match_0 motif logo (using the command chrombpnet footprints and make sure that the bias models footprint is closer to that of controls with no motif inserted - for examples look at FAQ )
| pattern | NumSeqs | cwm_fwd | cwm_rev | match0 | qval0 | match0_logo | match1 | qval1 | match1_logo | match2 | qval2 | match2_logo |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pos__0 | 9333 | |
|
TN5_2 | 1.708290e-06 | |
TN5_1 | 0.000020 | |
TN5_7 | 0.000469 | |
| pos__1 | 6211 | |
|
TN5_4 | 2.383960e-02 | |
TN5_5 | 0.023840 | |
TN5_8 | 0.041770 | |
| pos__2 | 3883 | |
|
TN5_3 | 1.530650e-01 | |
KLF4_MA0039.3 | 0.153065 | |
TN5_4 | 0.153065 | |
| pos__3 | 3813 | |
|
TN5_1 | 2.463760e-05 | |
TN5_2 | 0.000025 | |
TN5_3 | 0.000356 | |
| pos__4 | 2820 | |
|
TN5_3 | 1.633020e-03 | |
TN5_7 | 0.002835 | |
TN5_1 | 0.048250 | |
| pos__5 | 1938 | |
|
TN5_3 | 1.059630e-10 | |
TN5_4 | 0.000126 | |
TN5_5 | 0.000126 | |
| pos__6 | 1108 | |
|
TN5_3 | 2.597330e-07 | |
TN5_2 | 0.239226 | |
TN5_4 | 0.487056 | |
| pos__7 | 923 | |
|
TN5_3 | 1.846660e-06 | |
TN5_1 | 0.004382 | |
TBX21_TBX_3 | 0.035461 | |
| pos__8 | 743 | |
|
TN5_3 | 8.579450e-13 | |
TN5_4 | 0.001044 | |
TN5_5 | 0.001044 | |
| pos__9 | 398 | |
|
ZNF384_MA1125.1 | 5.112750e-02 | |
PRDM6_HUMAN.H11MO.0.C | 0.051127 | |
ANDR_HUMAN.H11MO.0.A | 0.204980 | |
| pos__10 | 304 | |
|
NFATC1_NFAT_1 | 1.000000e+00 | |
None | NaN | None | NaN | ||
| pos__11 | 213 | |
|
ZNF384_MA1125.1 | 5.872460e-03 | |
CPEB1_RRM_1 | 1.000000 | |
SRY_HUMAN.H11MO.0.B | 1.000000 | |
| pos__12 | 197 | |
|
LIN54_MA0619.1 | 1.000000e+00 | |
ZNF384_MA1125.1 | 1.000000 | |
ONECUT3_CUT_1 | 1.000000 | |
| pos__13 | 141 | |
|
MEF2A_MOUSE.H11MO.0.A | 2.159200e-01 | |
ZNF384_MA1125.1 | 0.215920 | |
ONECUT3_CUT_1 | 0.215920 | |
| pos__14 | 117 | |
|
TN5_7 | 4.013820e-05 | |
SOX8_HMG_3 | 0.751547 | |
SOX4_HMG_1 | 1.000000 | |
| pos__15 | 92 | |
|
FOXJ3_HUMAN.H11MO.0.A | 5.215270e-02 | |
FOXJ3_MOUSE.H11MO.0.A | 0.052153 | |
ONECUT3_CUT_1 | 0.052153 | |
| pos__16 | 76 | |
|
DNASE_2 | 2.335610e-01 | |
ZNF384_MA1125.1 | 1.000000 | |
SRY_MA0084.1 | 1.000000 | |
| pos__17 | 27 | |
|
TN5_3 | 5.018390e-01 | |
TN5_1 | 0.605895 | |
P53_HUMAN.H11MO.0.A | 0.622577 | |
| pos__18 | 25 | |
|
SRF_MADS_1 | 9.687150e-02 | |
SRF_MA0083.3 | 0.096871 | |
SRF_MADS_2 | 0.096871 | |
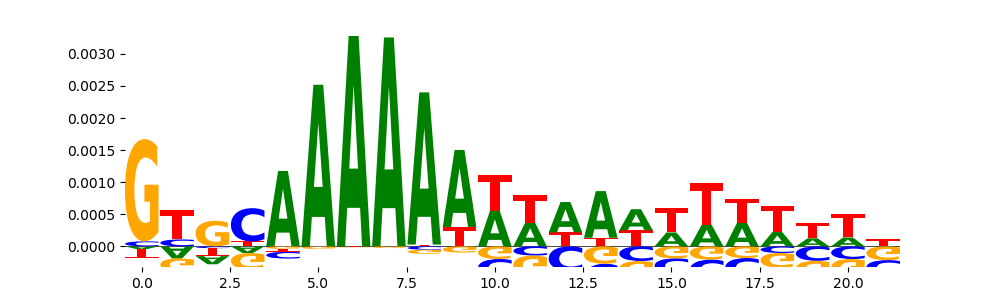
TFModisco motifs generated from counts contribution scores of the bias model. cwm_fwd, cwm_rev are the forward and reverse complemented consolidated motifs from contribution scores in subset of random peaks. These motifs should be free from any Transcription Factor (TF) motifs and should contain motifs either weakly related to bias motifs or random repeats. For each of these motifs, we use TOMTOM to find the top-3 closest matches (match_0, match_1, match_2) from a database consisting of both MEME TF motifs and heterogenous enzyme bias motifs that we have repeatedly seen in our datasets. The qvals should be high (> 0.0001) if the closest hit is a TF motif (i.e indicating that the closest match is not the correct match, this is also generally verifiable by eye and making sure the closest match looks nothing like the CWMs).
What to do if you find an obvious TF motif in the list?
Do not use this bias model as it will regress the contribution of the TF motifs (along with bias motifs) from the chrombpnet_nobias.h5. Reduce the bias_threshold_factor argument input to the chrombpnet bias pipeline or chrombpnet bias train command used in training the bias model and retrain a new bias model. For more intuition about this argument refer to the FAQ section in wiki.
What to do if you are unsure if a given CWM motif is resembling the match_0 logo for example?
Get marginal footprint on the match_0 motif logo (using the command chrombpnet footprints and make sure that the bias models footprint is closer to that of controls with no motif inserted - for examples look at FAQ )
| pattern | NumSeqs | cwm_fwd | cwm_rev | match0 | qval0 | match0_logo | match1 | qval1 | match1_logo | match2 | qval2 | match2_logo |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pos__0 | 14297 |  |
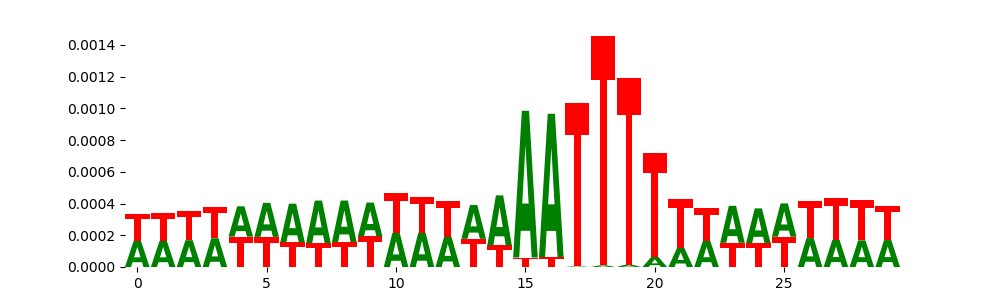 |
DNASE_2 | 2.038460e-01 | 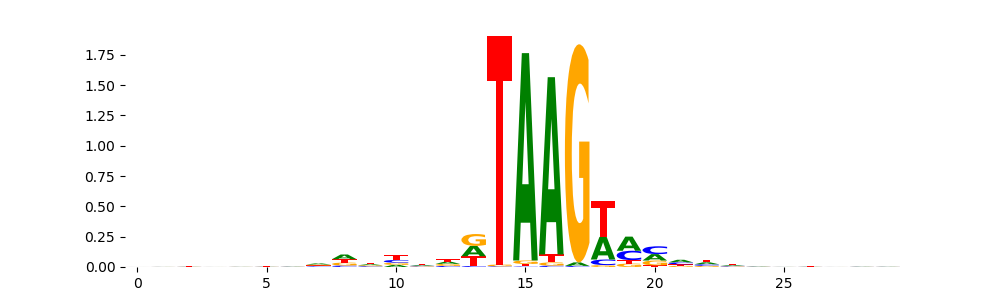 |
ZNF384_MA1125.1 | 2.319410e-01 |  |
LHX3_HUMAN.H11MO.0.C | 2.319410e-01 | 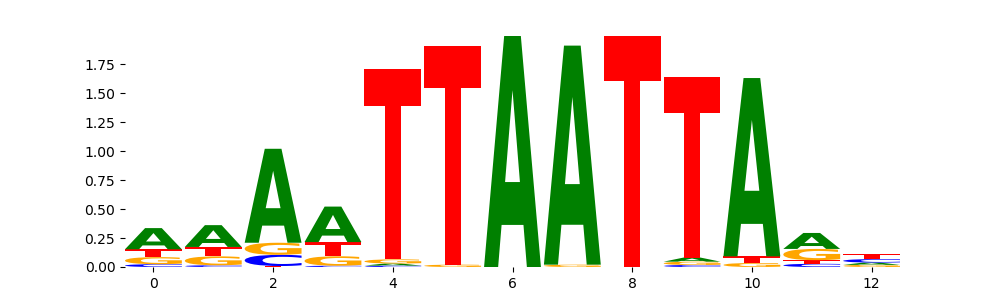 |
| pos__1 | 4373 | 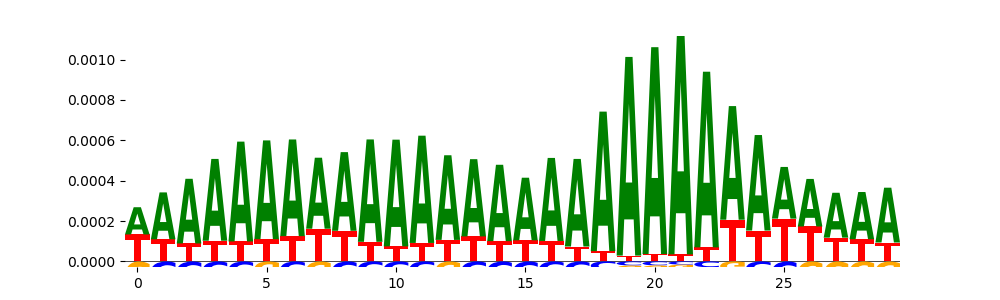 |
 |
ZNF384_MA1125.1 | 2.912580e-02 | |
PRDM6_HUMAN.H11MO.0.C | 2.912580e-02 |  |
STAT1_MOUSE.H11MO.0.A | 1.744390e-01 | 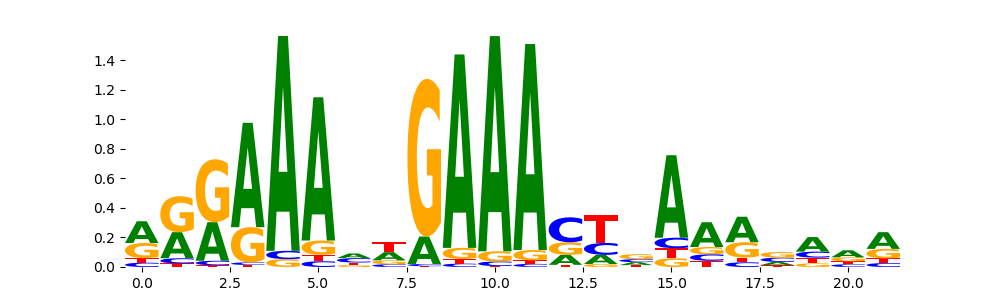 |
| pos__2 | 3792 |  |
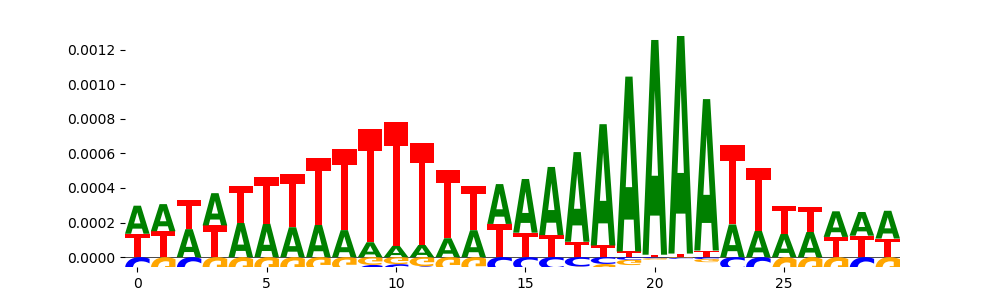 |
ZNF384_MA1125.1 | 2.822410e-03 | |
DNASE_2 | 2.583360e-02 | |
HOMEZ_HOMEZ_1 | 3.881610e-01 |  |
| pos__3 | 1876 | 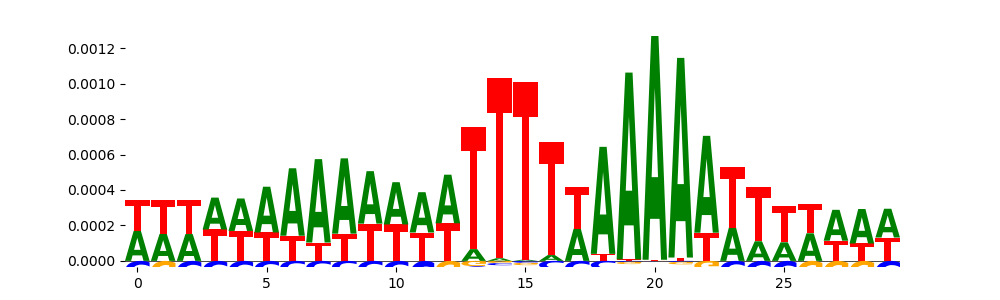 |
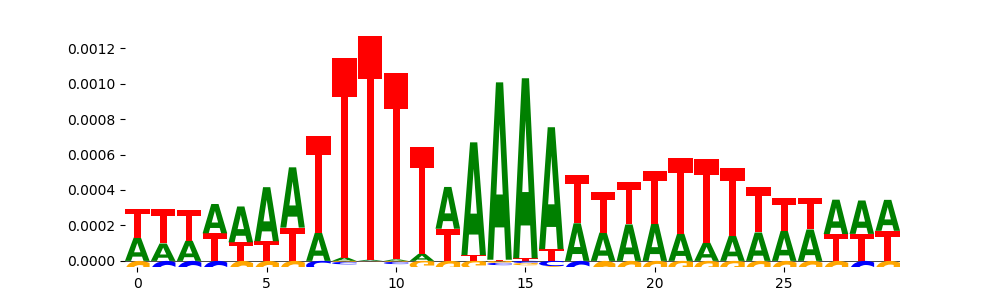 |
DNASE_2 | 3.285700e-02 | |
ZNF384_MA1125.1 | 1.000000e+00 | |
FOXD2_forkhead_1 | 1.000000e+00 |  |
| pos__4 | 1127 |  |
 |
DNASE_2 | 1.764790e-01 | |
CPEB1_RRM_1 | 1.764790e-01 |  |
IRF1_MA0050.2 | 1.764790e-01 | 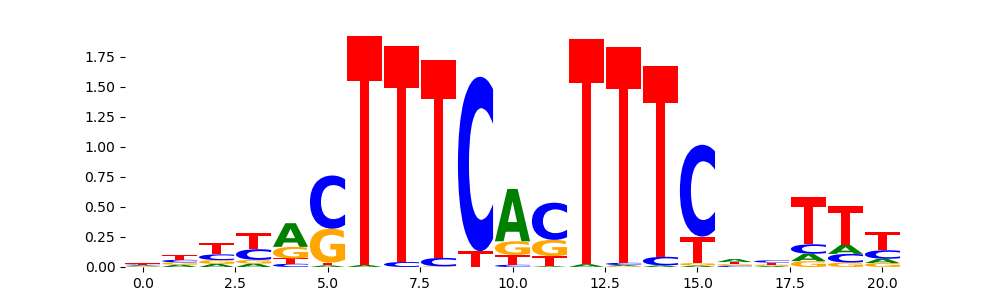 |
| pos__5 | 45 |  |
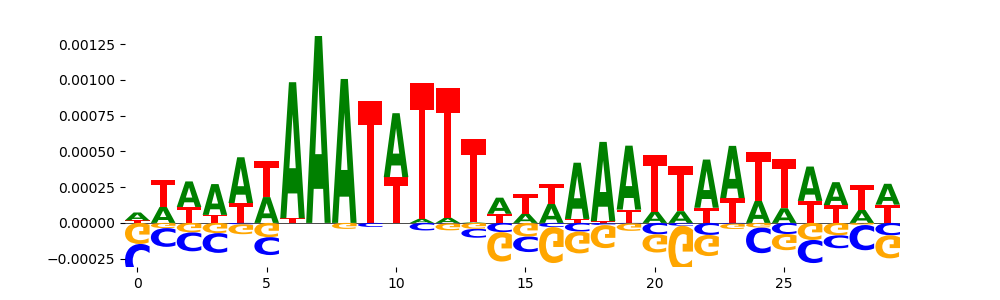 |
FOXD2_forkhead_1 | 4.332670e-02 | |
FOXD3_forkhead_1 | 1.387830e-01 |  |
PIT1_HUMAN.H11MO.0.C | 1.387830e-01 |  |
| pos__6 | 22 |  |
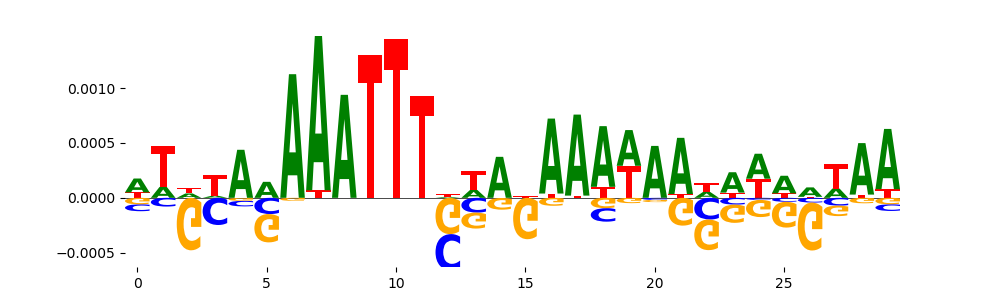 |
HSF1_MOUSE.H11MO.0.A | 1.725320e-02 | 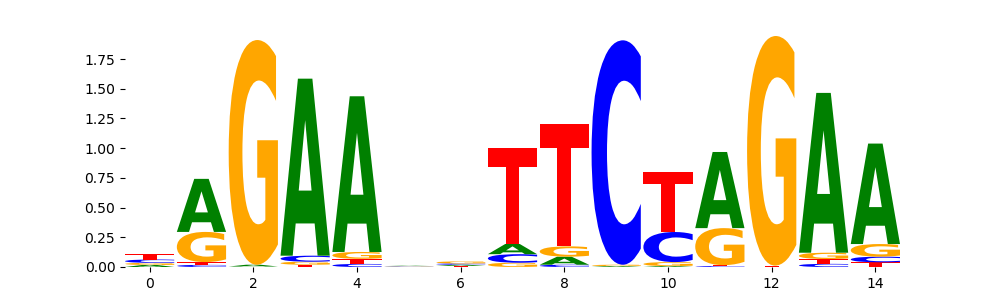 |
HSF1_HUMAN.H11MO.0.A | 2.822410e-01 | 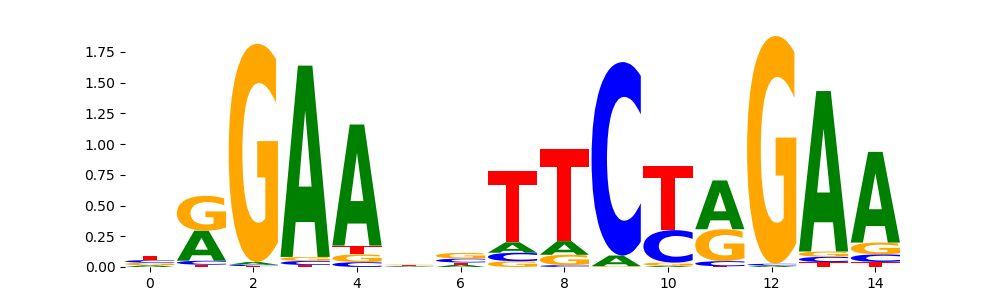 |
HSF2_HUMAN.H11MO.0.A | 2.822410e-01 | 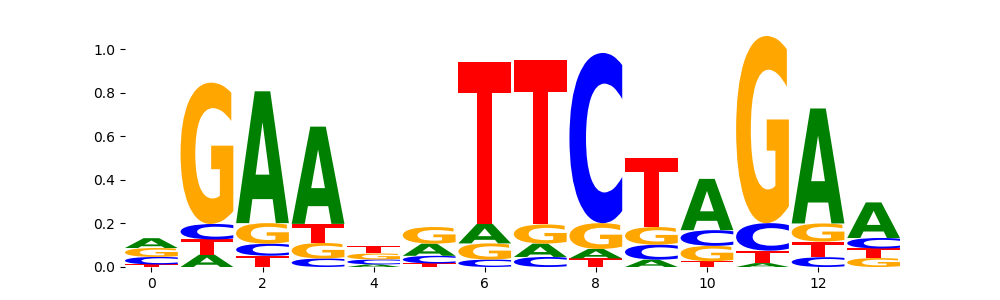 |