The best MEME-motif:
Total number of peaks (no FDR-cutoff) | = | 11486.0 |
Total number of reads | = | 17186481.0 |
Total number of UMIs | = | 17186481.0 |
Reads/UMIs | = | 1.0 |
Quality control results from FastQC for the input library.
The best MEME-motif: |
 |  |
Histogram of peak scores for all reported peaks. | Histogram of unique read 5'-end counts within peak borders (Reads pointing towards peak summit minus reads pointing away from peak summit). If UMIs were not used in the analysis, raw read counts are reported. |
 |  |
Histogram of UMI-counts per position on the sense strand. | Histogram of UMI-counts per position on the antisense strand. |
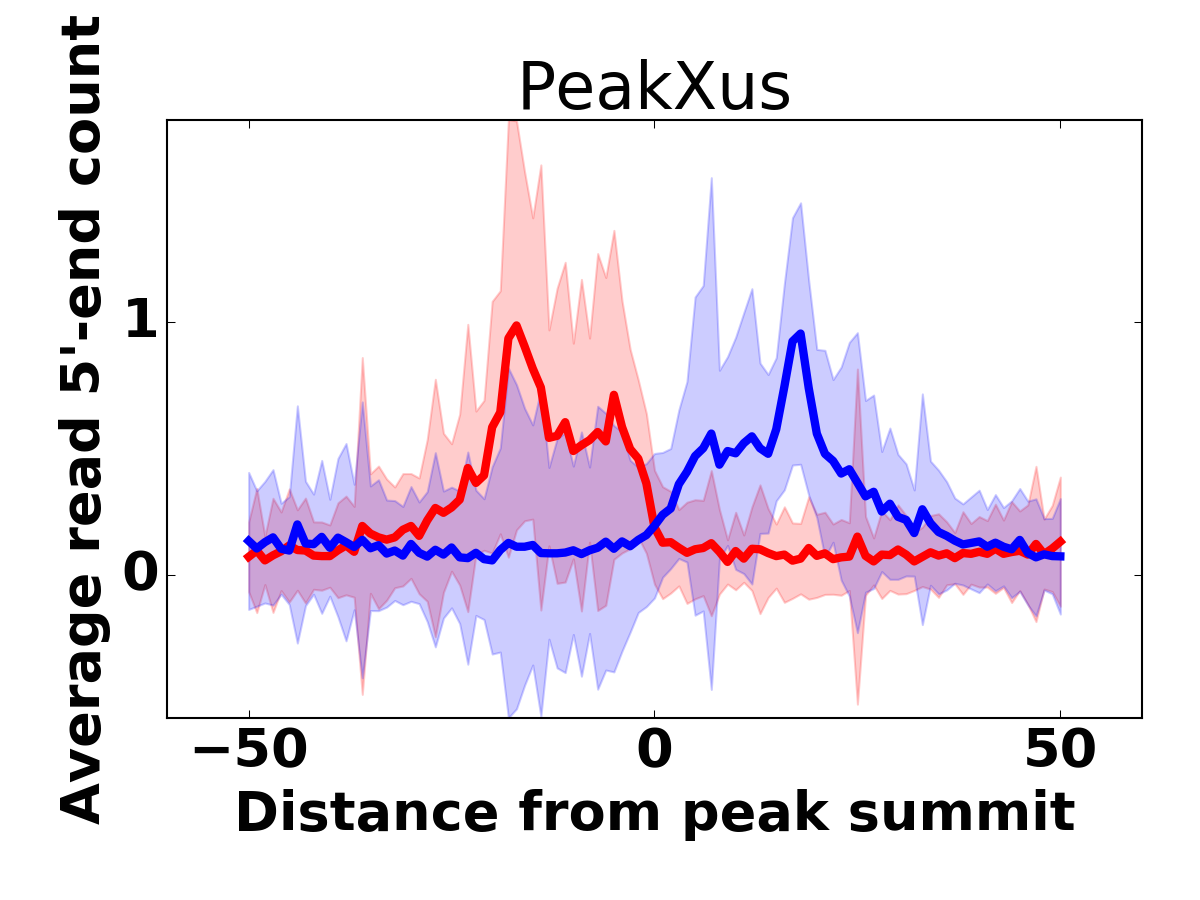 | 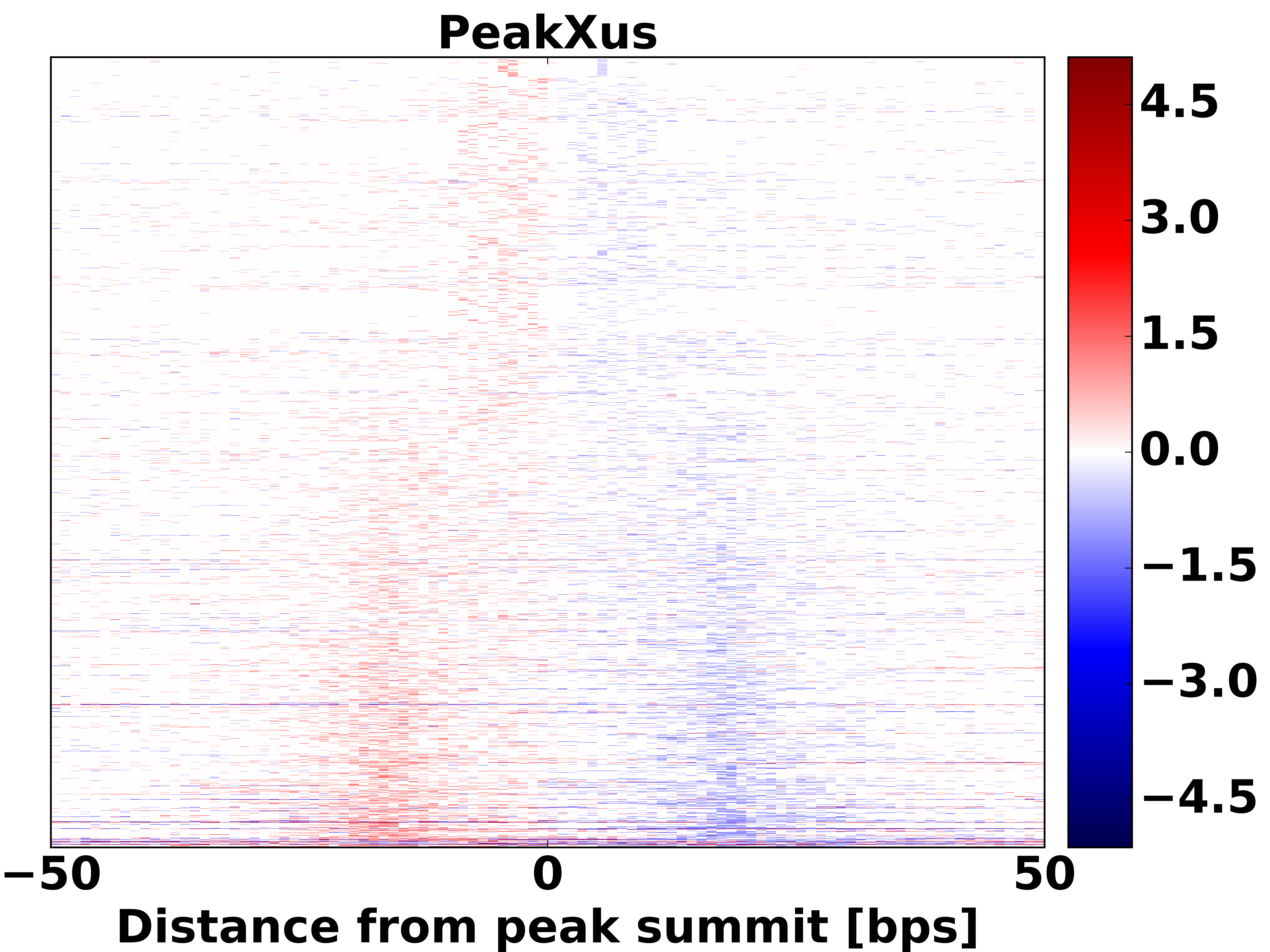 |
Average read 5'-end count around the top 1000 peaks. Red curve corresponds to read 5'-end counts on the sense strand, and blue on the antisense strand, respectively. | Heatmap presentation of the same peaks shown on the left. Red color marks the sense strand read 5'-end count, and blue the antisense strand, respectively. |
B2=None
N4=1000
N6=1000
S4=1
UMIs=None
adapters1=None
b3=0.05
blacklist='/oak/stanford/groups/akundaje/marinovg/genomes/blacklists/hg38.blacklist.bed'
center6=0
chromnames='/oak/stanford/groups/akundaje/marinovg/genomes/hg20/hg38-male.chrom.sizes'
e4='PeakXus'
fastq='ZNF302-SRR5197107.hg20-male.36mers.unique.bam'
genome2=None
k4=4
l3=5
l6='PeakXus'
l_l3=60
m4=10
m5=20
m6=100
matrixhits=None
maxw7=50
minw7=4
ml4=17
ms4=-8
n3=1
n4=1
o5=1
outdir='ZNF302-SRR5197107/peakXus/'
p3=1.0
p7=1
q2=20
s3=1
skip0=0
t2=1
title5='PeakXus'
verbosity=1
w3=5
w4=100
wg7=None)Testing that input files exist...done!
If you use PeakXus in your work, please cite:
Hartonen, T., Sahu, B., Dave, K., Kivioja, T., & Taipale, J. (2016). PeakXus: comprehensive transcription factor binding site discovery from ChIP-Nexus and ChIP-Exo experiments. Bioinformatics, 32(17), i629-i638.